We have developed the project for the [Genome Antibody Discovery Drugs] based on the technology of mouse monoclonal antibodies. 12 lead antibodies for cancer treatment have been identified and that number is increasing. If all of these antibodies are humanized and engineered to be multifunctional to kills cancer cells, they will provide revolutionary progress for the treatment of advanced cancer.
An antibody is an extremely stable protein in our body. However, the engineering and alteration of the amino acid sequence of the antibody generates the following problems during the humanization or pre-targeting process;
(1) The expression of functional recombinant protein will be difficult, as the folding process changes significantly and aggregation occurs easily with the engineered antibody.
(2) Decreased affinity for antigen and change in the disposition.
(3) Produces immunogenicity in the human body from alteration of amino acid sequence.
In order to slove these problems, advanced computer technology provides an advantage. With the progress of computing power, the improvement of the function of engineered antibodies became possible by computer simulation using molecular dynamics.
An antibody is an extremely stable protein in our body. However, the engineering and alteration of the amino acid sequence of the antibody generates the following problems during the humanization or pre-targeting process;
(1) The expression of functional recombinant protein will be difficult, as the folding process changes significantly and aggregation occurs easily with the engineered antibody.
(2) Decreased affinity for antigen and change in the disposition.
(3) Produces immunogenicity in the human body from alteration of amino acid sequence.
In order to slove these problems, advanced computer technology provides an advantage. With the progress of computing power, the improvement of the function of engineered antibodies became possible by computer simulation using molecular dynamics.
design engineered antibodies
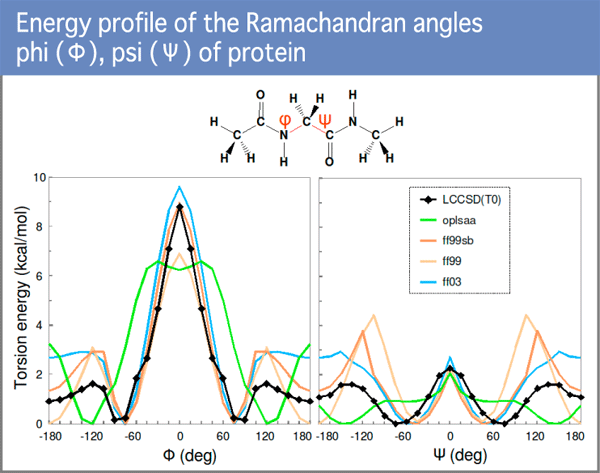
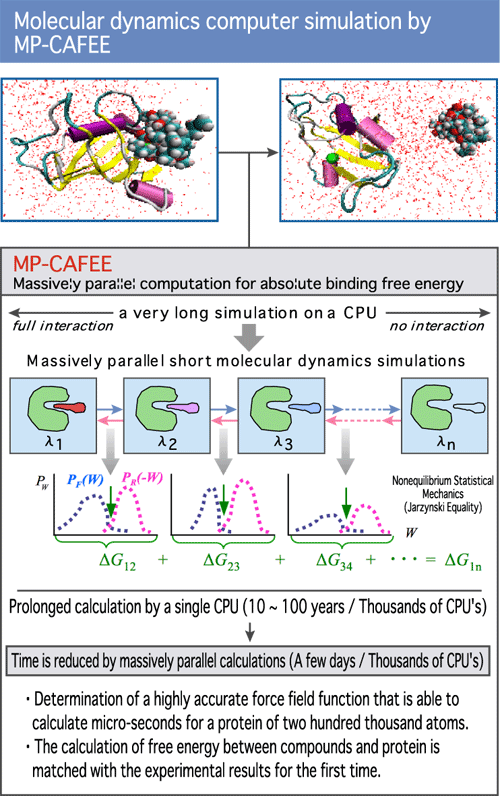
Dr. Fujitani, the leader of the molecular dynamics design team at the University of Tokyo, is the world’s authority on developing algorithms to calculate solvated energy in protein and the prediction of protein-ligand binding free energy. He created highly accurate force fields, for discrepancies in AMBER (Assisted Model Building with Energy Refinement) parameters which has been considered the gold standard. The figure above shows the energy profile of the Ramachandran angles phi (Φ) and psi (Ψ) (Diagram 7). The black line indicates LCCSD (linearized coupled-cluster)-results and the other lines indicate the profile of the standard protein force fields. This disparity is attributable to the low quality of the standard protein force fields.
The new field of MP-CAFFEE, developed by Fujimoto, matches the black line (2009, Phys Rev). GROMACS version 4.1, which will include MP-CAFEE functionality, has been released in 2010 by developers in Sweden. With GROMACS 4.1, efficient, large-scale parallel simulations will become available to the public. (Diagram 8)
To increase the binding affinity of an antibody for its antigen, it is essential to optimize the location of six of its CDR loop's amino acid sequences and antigen amino acids.
Dr. Tsuchiya (The University of Tokyo), co-investigator of the molecular dynamics design team, is developing methods to engineer humanized protein from non-human species using human protein amino acid sequences. His research greatly contributes to the design modification of streptavidin amino acid sequences. (Diagram 9)
It is crucial to use both structure and sequence based predictions for delicate protein assemblies.
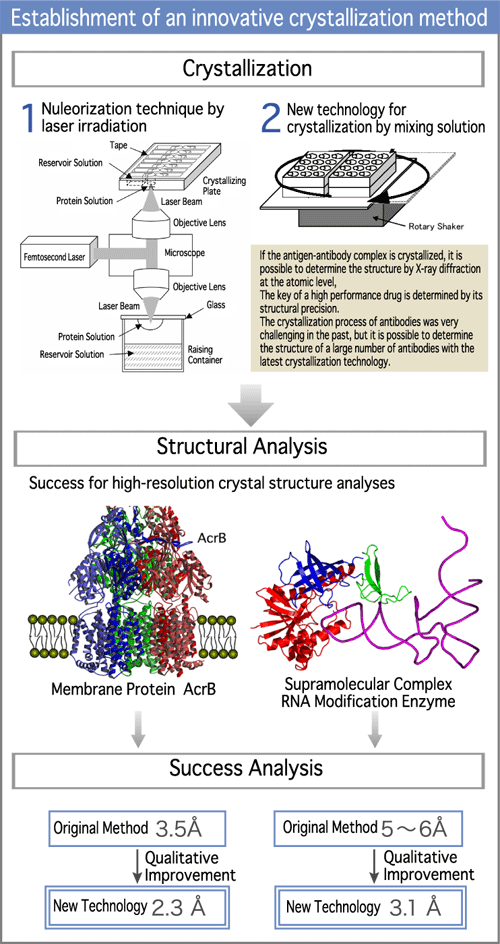
Dr. Inoue, the leader of the crystal analysis team is specialized in crystal engineering. He has produced high-quality crystals of the antigen-antibody complex.
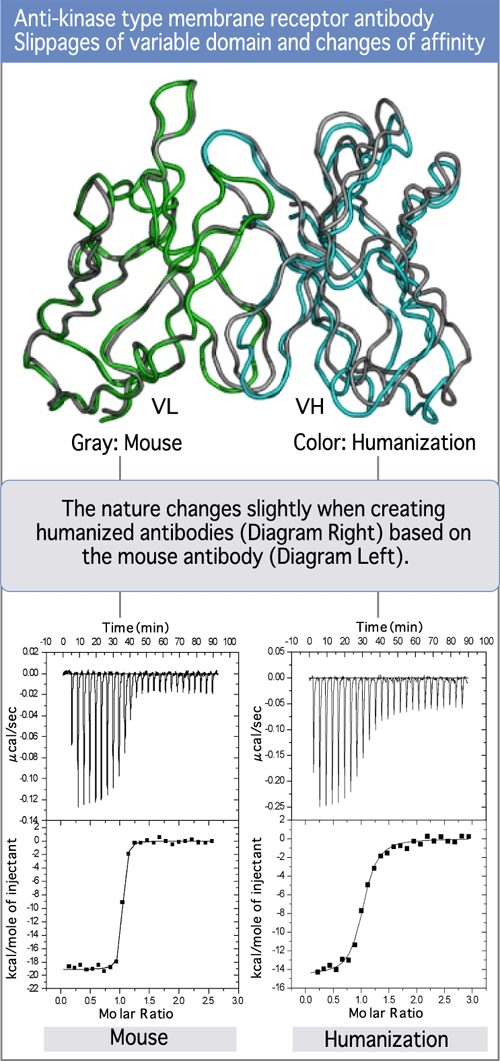
Dr. Tsumoto, the leader of the thermodynamics analysis team at the Institute of Medical Science, University of Tokyo, is experienced in expressing and re-folding scFv's. He uses microcalorimeters to analyze antigen-antibody complexes and assess the entropy changes. (Diagram 10)